4.3.5. Create Bonds operator¶
The CreateBonds operator is used to specify ranges of distances for various types of atoms and use those ranges to create bonds. The default behavior of this operator is to create a bonds between a Hydrogen and any other species if the atoms are separated by a distance between 0.4 and 1.2 units (e.g. Angstroms) and between a pair of atoms (not including H) if they are between 0.4 and 1.9 units apart. This works well for organic molecules. However, in Figure 4.20, the default distances were not useful. In this case, the values were changed to create a bond including H for distances between 0.4 and 1.5 units and for other species between 0.4 and 2.5 units. Figure 4.21.
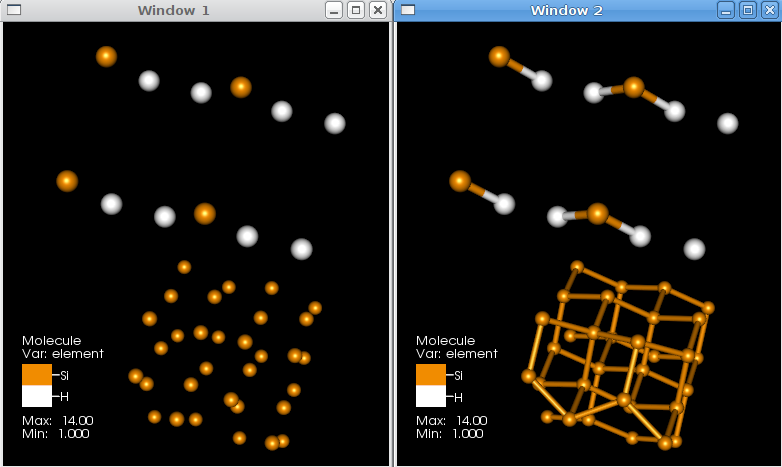
Fig. 4.20 Bonds created with different bonding distances¶
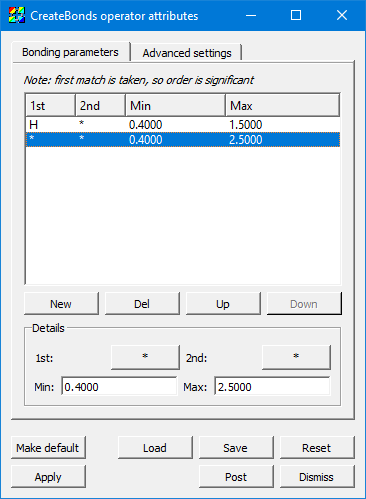
Fig. 4.21 CreateBonds bonding parameters¶
4.3.5.1. Setting the Bonding parameters¶
The Bonds list contains the bonding pair specifications to algorithm. Each row contains the species 1st and 2nd, and the Min and Max distance which could be considered a bond.
Note:
A “*” matches any species.
It does not matter which species is 1st and which is 2nd. The bonds are not unidirectional.
The first match in the list is taken, even if later lines also match, which allows you to specify more specific rules above less-specific rules.
For example, if the first line is “H”, “*”, “0.4”, “1.2”, this specifies that the algorithm should create a bond between two atoms if either one is Hydrogen and the distance between them is between 0.4 and 1.2 spatial units.
As a follow-on to this example, suppose the second line is “*”, “*”, “0.4”, and “1.9”. Now suppose two atoms exist, H and O, and they are separated by a distance of 1.5 units. Because one is H, it will match the first line, determine the distance is too great (since it’s greater than 1.2), and so it will not create a bond between this atom pair. Since this atom pair matched the first line, the second line is not considered, even though the atom pair matches its criteria.
Just below the actual bonding rules list are several buttons: The New button creates a new rule, and Del deletes it. Up moves the currently selected rule up in the list, and Down moves the currently selected rule later in the list. Recall that the order of rules matters because only the first match is considered.
The Details section contains controls to set the values for a rule. The 1st and 2nd controls pop up the species selection widget shown in Figure 4.22.
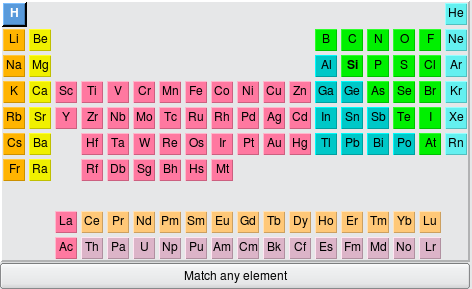
Fig. 4.22 The element selector¶
To get a wildcard which will match any type of atom, choose Match any element at the bottom; it is selectable just as any individual species in the periodic table.
Also, note that there is the possibility for some hinting to help guide your selection to the viable types of atoms. (This depends on conditions like file format support.) For example, in this screenshot, the H and Si elements are in boldface, since the file contains only those types of atoms. The Min and Max fields are standard text widgets.
4.3.5.2. Advanced settings¶
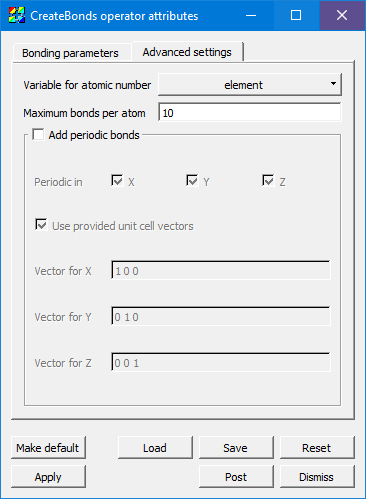
Fig. 4.23 The advanced settings tab¶
Variable for atomic number defaults to element as per the convention, but it can be set to any integral variable corresponding to the atomic number of each atom.
If you specify the wrong distance, each atom might try to bond to many other atoms. To keep an error like this from causing a severe hit to memory and performance, Maximum bonds per atom will stop the process before it gets out of hand. The default value is 10, and it could safely be set lower in many cases, but it is user-settable for unusual cases where >10 bonds are needed on some atoms.
When Add periodic bonds is checked, this will make the algorithm see if an atom would bond with an atom past a periodic boundary edge, and add a dangling bond in that case. Checking this setting will enable the Periodic in X,Y,Z controls as well as the controls for Unit cell vectors.
Periodicity in X, Y, Z can be selected independently (or none).
Some file formats specify the vectors for the unit cell (sometimes called “direct lattice” vectors) containing the molecular data in the file. If they are present and Use provided unit cell vectors is checked, then it will use those values instead of the ones specified in this window.
Vector for X, Y, and Z controls the actual vectors describing the amount to displace in each of the three axes.
4.3.5.3. Examples in use¶
See Molecular data features for examples of the CreateBonds operator in use with the Molecule Plot.